
Psychedelics on prescription: How far does Trump's mental health Executive Order actually go?
Psychedelics at the Policy Frontier: What Trump’s Mental Health Executive Order Means for Biotech Investors

On 18 April 2026, President Trump signed an executive order titled “Accelerating Medical Treatments for Serious Mental Illness,” directing federal agencies to fast-track the development, regulatory review, and rescheduling of psychedelic compounds for treatment-resistant psychiatric conditions. The order arrived with the full staging of a political moment: signed in the Oval Office alongside HHS Secretary RFK Jr., FDA Commissioner Marty Makary, podcaster Joe Rogan, and several military veterans. That staging matters for investors, because it tells you something about who is driving this and why.
")
President Trump at the signing of the Executive Order with Jay Bhattacharya, Marty Makary, Robert F. Kennedy Jr.,Joe Rogan, W. Bryan Hubbard, Morgan Luttrell & Marcus Luttrell
The policy ambitions are real. The commercial implications are significant. But this is a more complicated picture than the initial stock rally suggests, and investors who treat this as a simple green light are reading it wrong.
The Problem Being Addressed
The order’s statutory foundation rests on two well-documented crises. Over 14 million American adults currently meet criteria the for serious mental illness (SMI), defined as a diagnosable mental, behavioural, or emotional disorder that substantially interferes with daily functioning. Of those, roughly 8 million are on prescription medication, and a meaningful proportion do not achieve sustained remission with existing therapies.

Suicide rates rose 37% between 2000 and 2018, peaked again in 2022 following the height of the COVID-19 pandemic, and remain disproportionately concentrated in the veteran population: over 6,000 veteran suicides per year for more than two decades, at a rate more than twice that of the non-veteran adult population.
Mental illness remains widespread, affecting nearly 1 in 4 (23.4%) of U.S. adults in 2024 and 17.6% of veterans in 2023, according to the National Institute of MEntal Health. These are not marginal numbers. They define a large, underserved patient population with unmet clinical need and, consequently, a substantial commercial addressable market.

What the Order Actually Does
The EO contains five operative sections, each directing specific agencies to take specific actions. Reading the text carefully matters here, because the gap between the headline and the mechanism is wider than the press coverage implies.
Section 2 directs the FDA Commissioner to issue Commissioner’s National Priority Vouchers (CNPVs) to “appropriate” psychedelic drugs that have received Breakthrough Therapy designation and meet the criteria of the CNPV programme. CNPVs, introduced in June 2025, compress review timelines from the standard six to ten months down to one to two months. The FDA moved immediately: on 24 April 2026, it issued CNPVs to three products. Compass Pathways received one for COMP360, its synthetic psilocybin for treatment-resistant depression; the Usona Institute received one for its psilocybin candidate for major depressive disorder; and Transcend Therapeutics (now acquired by Otsuka for USD 700 million) received one for its methylone-based PTSD treatment. Compass CEO Kabir Nath described the voucher as providing “some more momentum” toward potential approval, while noting it would not represent a “huge acceleration” for Compass specifically, given its existing rolling submission timeline.
Section 3 requires HHS to allocate at least USD 50 million from existing funds through the Advanced Research Projects Agency for Health (ARPA-H) to match state government investments in psychedelic research programmes. This provision appears to have been written in direct response to Texas, which in 2025 committed USD 100 million to a publicly funded ibogaine research consortium after failing to secure a required private sector match.
Section 4 directs HHS, FDA, and the VA to sign data-sharing memoranda to pool clinical trial data, increase trial participation, and generate real-world evidence, prioritising compounds with Breakthrough Therapy designation.
Section 5 directs the Attorney General to initiate rescheduling review for any product containing a Schedule I substance that has successfully completed Phase 3 trials, so that rescheduling can proceed as quickly as practicable following FDA approval.
Section 2(b) also instructs the FDA and DEA to establish a pathway for eligible patients to access investigational psychedelic drugs, including ibogaine compounds, under the Right to Try Act. The same section requires Schedule I handling authorisations for treating physicians and researchers.
The Ibogaine Question
Ibogaine is explicitly named twice in the EO text, which is notable given where the science actually sits. The compound, derived from Tabernanthe iboga, a shrub native to central Africa, has attracted significant advocacy from the veteran community and from figures including Joe Rogan and former Texas Governor Rick Perry. The political case for it is clear. The clinical case is not. Ibogaine carries documented cardiac toxicity risk, specifically QT prolongation, which has historically made the FDA resistant to approving research.
Despite the political attention, the FDA did not include ibogaine in the initial CNPV recipients. The agency did, however, grant the first Investigational New Drug (IND) clearance for DMX-1001, an oral noribogaine derivative developed by AtaiBeckley subsidiary DemeRX NB, intended for alcohol use disorder. DemeRX’s CEO Deborah Mash has stated that noribogaine does not carry the same cardiac safety profile as the parent compound, and the distinction between ibogaine and its derivatives matters considerably for any regulatory pathway analysis. The EO’s ibogaine emphasis is best understood as politically motivated: honouring a constituency while directing the actual commercial acceleration toward compounds with more advanced and less safety-compromised profiles.
Market Response and Named Beneficiaries
The initial market reaction on 21 April was sharp. At least six publicly traded psychedelics biotechs saw their stocks rise, several by high single-digits or more. Compass Pathways (CMPS) climbed 53% from Friday’s close to its Monday high. AtaiBeckley (ATAI) rose 36%. GH Research (GHRS) gained 34%. Enveric Biosciences (ENVB) nearly tripled, rising 198%. By 24 April, following CNPV confirmation, Compass shares were trading approximately 2% up on the day, reflecting some consolidation after the initial spike.
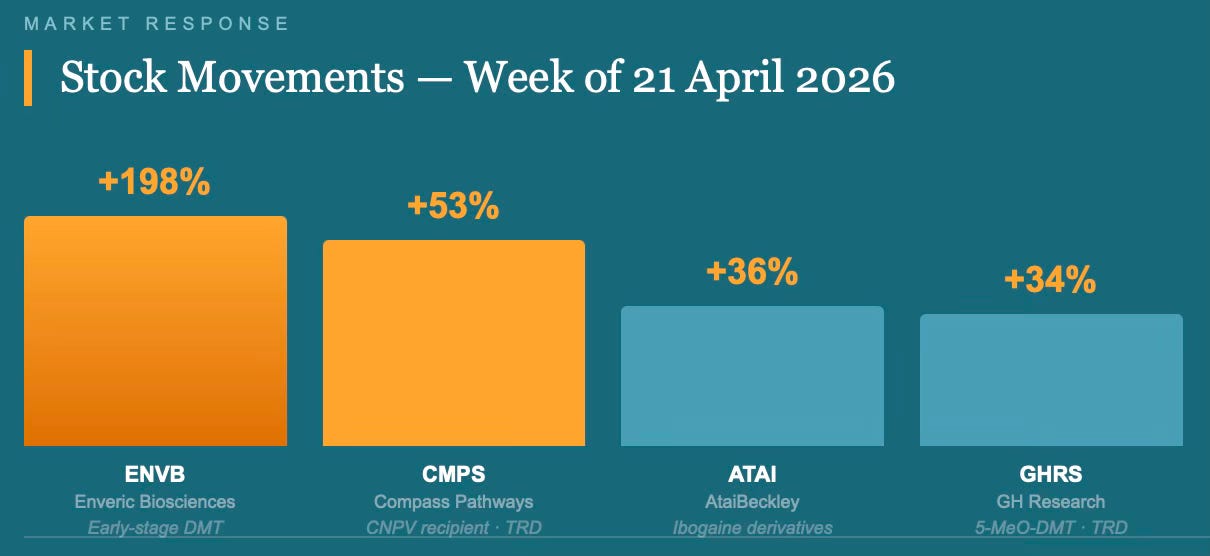
Analyst reaction was structured around two themes. Jefferies analyst Andrew Tsai called the EO “an official stamp of validation to the class” and described the combination of regulatory, legal, and deal-making momentum as making psychedelics “an investable space.” RBC analyst Brian Abrahams described the order as “a substantial step towards diminishing regulatory risk in this emerging class of therapies, enabling investor comfort,” and identified the CNPV programme and the expedited rescheduling provision as the two most direct mechanisms for accelerating potential revenue generation. Oppenheimer analysts termed the development “a structural inflection for the U.S. psychedelics sector.” Stifel’s Paul Matteis identified Compass and Definium Therapeutics (DFTX), formerly MindMed, as the two most obvious CNPV beneficiaries. Definium holds a synthetic LSD derivative, DT120, in Phase 3 for generalised anxiety disorder and major depressive disorder.
The broader deal-making context reinforces the investment case. The sector has been attracting strategic interest at scale: Otsuka acquired Transcend for USD 700 million in March 2026; AbbVie acquired Gilgamesh Pharmaceuticals in a deal that generated significant M&A momentum for the space. Johnson & Johnson’s Spravato (esketamine), a ketamine-derived treatment for treatment-resistant depression, has delivered commercial proof-of-concept for the delivery model, providing a precedent that analysts cite when building the bull case for psilocybin-based therapies.
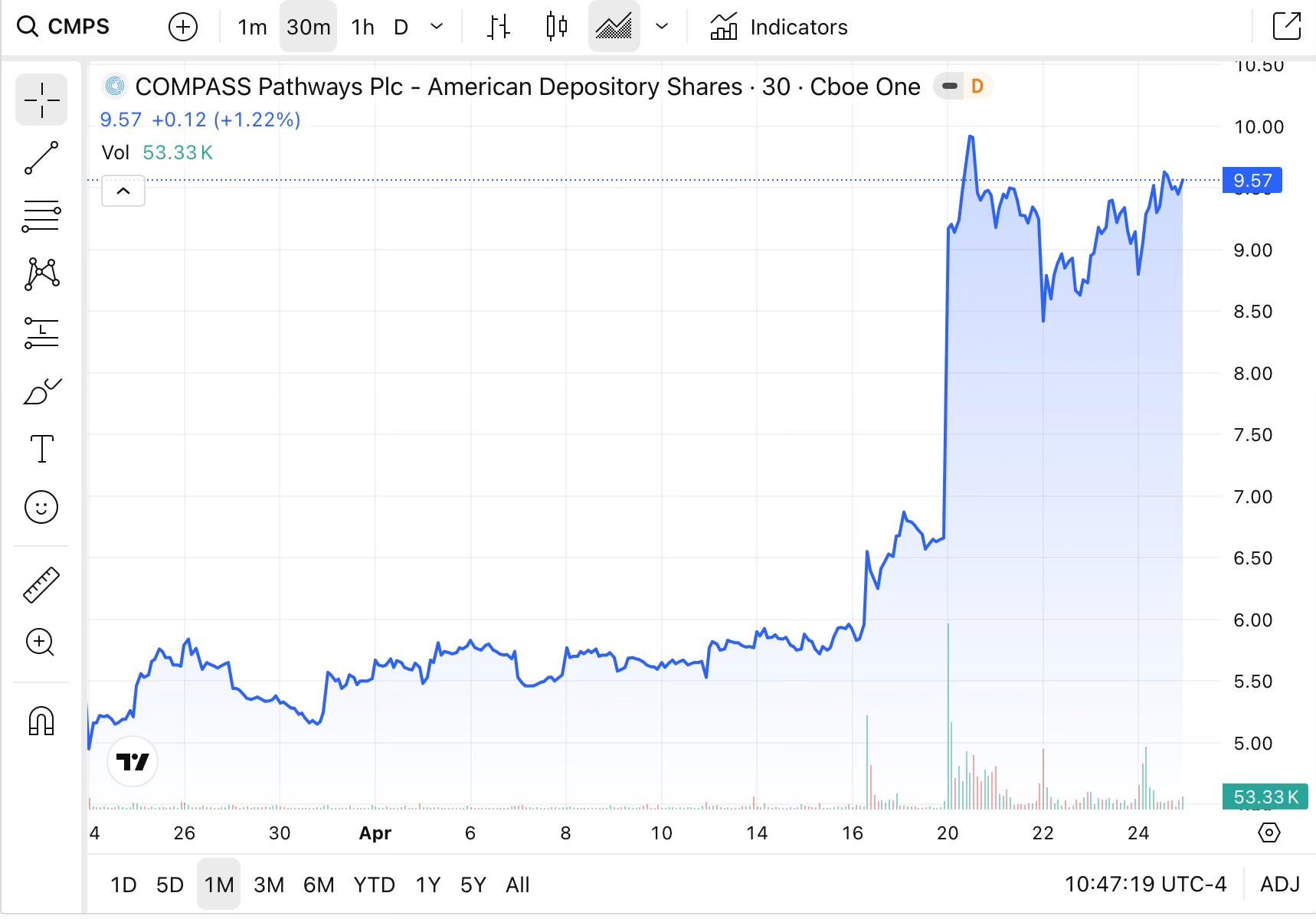
Created with Trading view
Federal Architecture and Agency Responsibilities
The EO assigns responsibilities across four agencies with distinct mandates and, in some cases, distinct institutional cultures that will shape implementation pace.
The FDA sits at the centre of the order’s operative provisions. Commissioner Makary was present at the signing and has publicly signalled alignment with the administration’s acceleration agenda. The CNPV programme already existed; the EO instructs Makary to apply it to this compound class. The “shall” language in Section 2(a) sounds mandatory, but as Harvard Law’s I. Glenn Cohen notes, the provision adds “in accordance with the criteria of the National Priority Voucher Program,” which preserves considerable FDA discretion. The more significant signal may be political: the EO implicitly reverses a reported White House decision in February 2026 that had blocked COMP360 from CNPV inclusion.
HHS, through ARPA-H, is responsible for the USD 50 million allocation. The critical phrase is “from existing funds,” which does not constitute new appropriations and is explicitly conditioned on “availability of appropriations” under Section 6(b). In the context of federal-scale clinical trials for Schedule I compounds, USD 50 million is modest. Marks notes it “might not go very far” in FDA-sanctioned clinical research, though it could leverage additional private sector investment.
The DEA’s role is twofold: establishing Right to Try access pathways and initiating rescheduling reviews. On rescheduling, Harvard Law’s Mason Marks and Professor Rob Mikos have both noted that the DEA is already statutorily required to reschedule drugs within 90 days of FDA approval, making Section 5 largely redundant as an acceleration mechanism. Presidents cannot compel rescheduling; the EO can only request review. This is an important structural limitation for investors modelling timelines around scheduling reform.
The VA’s role is primarily collaborative: data sharing and trial participation. Given that veterans represent the most politically salient segment of the target patient population, and that VA-generated real-world evidence could be substantial, this provision has longer-term significance for the evidence base even if it does not produce immediate commercial impact.
Patient and Clinical Implications
For patients with treatment-resistant SMI, the order represents a genuine advance in access architecture, with important caveats. The Right to Try pathway, if the FDA and DEA implement it effectively, would allow eligible patients to access investigational psychedelic drugs before approval, provided those compounds have met Phase 1 safety requirements. That is materially different from the current situation, where expanded access exists but requires case-by-case application and institutional cooperation.
The delivery model question is unresolved and clinically significant. Cohen’s analysis is sharp here: the kind of Risk Evaluation and Mitigation Strategy (REMS) the FDA attaches to any eventual approval will heavily determine cost, accessibility, and the clinical framework. A REMS resembling the existing ketamine clinic model produces a very different access landscape to one that permits GP-level prescribing. The EO is silent on this. Reimbursement is equally absent from the text, despite being the dominant determinant of real-world patient access post-approval.CMS Administrator Dr. Oz has noted awareness of cost issues, but no reimbursement framework has been proposed.
For healthcare providers, particularly psychiatrists and addiction medicine specialists, the order signals a coming demand surge in a clinical area requiring specialised training in psychedelic-assisted therapy administration and monitoring. The infrastructure does not currently exist at scale.
Criticisms and Structural Concerns
The most substantive criticism is not that the EO accelerates psychedelic development, but that the mechanism of acceleration raises legitimacy questions that could harm the field.
Holly Fernandez Lynch, Associate Professor of Medical Ethics and Law at the University of Pennsylvania, described the White House directing the FDA toward specific products as “a pretty egregious step” and characterised the level of politicisation as “pretty extreme.” Her specific concern is the perception problem: “How terrible would it be for products that are really good to have a hanging cloud over them that maybe it was just politics that supported their approval.” She also noted that rapid reviews are resource-intensive and will deprioritise other applications, and that the FDA should make those trade-off decisions independently.
The account of Joe Rogan texting Trump and receiving a commitment to “let’s do it” on FDA approval, relayed at the signing ceremony, is not reassuring from a regulatory integrity standpoint. Fernandez Lynch was direct: “That level of executive influence over specific product decisions is not acceptable.”
Cohen raises a related concern about the CNPV signal being a “mixed blessing” for first-mover companies: a priority review voucher may signal to payers that the evidentiary bar was lower than standard, potentially complicating reimbursement negotiations precisely for the drugs that most need payer support to drive commercial uptake.
The ibogaine provisions present a specific regulatory integrity risk. The compound’s cardiac toxicity profile, documented concerns about long QT syndrome, and limited data place it outside standard Right to Try eligibility requirements, which mandate completion of Phase 1 trials. Marks notes directly that ibogaine has arguably not met the basic safety requirements that the Right to Try statute requires. Naming it twice in the EO text, and centralising it in the political narrative, creates pressure on the FDA to approve a Right to Try pathway for a compound that may not clinically warrant one. Moreover, meeting phase 1 outcomes is not the end of the safety debate. Medications can be found to have further safety issues when trialled in larger populations in Phase 2 and 3 trials or even at commercial rollout stages.
The HALT Fentanyl Act, signed in 2025, did meaningfully reform research pathways for Schedule I substances, including psychedelics, making this EO’s research provisions easier to operationalise. That is genuine legislative infrastructure. But the tension between political momentum and evidentiary rigour is real, and investors should factor reputational and regulatory backlash risk into their models.

Policy Landscape and Fit
This order does not arrive in a vacuum. It builds on a sequence of policy moves: Trump’s first-term Right to Try Act, which established the pre-existing legal architecture the EO now extends to psychedelics; an EO on increasing medical marijuana and cannabidiol research signed in December 2025; and bipartisan Congressional interest in psychedelic therapy legislation. The DOJ’s announcement on 23 April 2026 of relaxed marijuana scheduling rules signals a broader shift in federal drug policy that extends beyond any single executive action.
The order is also consonant with growing institutional engagement from Stanford, Harvard, Johns Hopkins, and Imperial College London, all of which have active psilocybin research programmes. That academic credibility matters for investor confidence, particularly after the FDA’s 2024 rejection of MDMA-assisted therapy from MAPS/Lykos created a significant setback for the sector and raised questions about how the FDA would handle psychedelic NDAs. The CNPV awards to Compass, Usona, and Transcend are a partial answer to that question.
The Investor Calculus
The near-term investment thesis is clearest for companies with the most advanced clinical profiles and existing Breakthrough Therapy designation. Compass Pathways is the most proximate commercial opportunity: COMP360 has positive Phase 3 data for treatment-resistant depression, a rolling NDA submission underway, and a CNPV that compresses its review window. The company is targeting a Q4 2026 submission completion and has said it aims to be “launch-ready by end of 2025.” Definium’s DT120, an LSD derivative with Phase 3 data in anxiety and depression, is the second most proximate beneficiary. H.C. Wainwright analyst Patrick Trucchio suggested in February that both COMP360 and DT120 could be approved by 2027; the CNPV advances that timeline.
Smaller names, including GH Research [GHRS] (5-MeO-DMT for treatment-resistant depression) and AtaiBeckley [ATAI] (ibogaine derivatives and nasal spray psilocybin), are further from approval but have benefited from the sector re-rating. Enveric Biosciences, whose DMT analogue is still in early development, nearly tripled on the news, which is a valuation that requires considerable caution.
The risks are structural rather than speculative. Reimbursement frameworks do not yet exist. Delivery model uncertainty, driven by unresolved REMS questions, creates cost and scalability unknowns. The perception problem is real and could affect payer willingness to cover expedited approvals and patient willingness to engage in a novel treatment modality. Then there is the political nature of the catalyst itself: administrations change, executive orders can be revoked, and the current policy environment is not a permanent feature of the landscape although in this case I don’t see this is a particularly likely risk given the potential backlash with veterans and mental health advocate groups.
The most durable investment thesis is not the approval timeline acceleration alone but the structural shift in how the FDA, DEA, and HHS are now institutionally positioned relative to this compound class, combined with independent evidence of commercial viability through Spravato’s sales performance and the M&A activity from Otsuka, AbbVie, J&J, and Lilly. The EO is a political event sitting on top of a genuine clinical and commercial inflection.

Conclusion
The executive order on “Accelerating Medical Treatments for Serious Mental Illness” is a meaningful regulatory event for the psychedelic therapeutics sector, and the FDA’s six-day response in issuing CNPVs to three products demonstrates that the administration is following through on the order’s directives with speed unusual by federal standards.
For biotech investors, the actionable implication is differential. Compass and Definium are the clearest direct beneficiaries, with CNPV-accelerated NDA timelines and robust Phase 3 datasets. The wider sector re-rating reflects both the EO’s signalling value and a broader policy shift that increases the investability of this compound class. The speculative end of the market, particularly early-stage names with thin clinical data, should be treated as trading the news rather than the fundamentals.
The substantive limitations are real. USD 50 million in matched ARPA-H funding from existing appropriations is not transformative research investment. The rescheduling provisions add minimal acceleration beyond what statute already requires. The ibogaine political narrative has created expectations that the clinical evidence does not yet support. And the absence of any reimbursement framework means approval, when it comes, will not automatically translate into access.
The patients who need these treatments most, those for whom SSRIs have failed, for whom years of treatment have not produced remission, are watching this carefully. The order is a welcome step in the right direction. Whether it produces durable, accessible, reimbursed care at population scale depends on what follows. That story is still being written.
Sources: White House EO text, 18 April 2026; White House Fact Sheet, 18 April 2026; Reuters, 20 and 24 April 2026; BioPharma Dive, 21 April 2026; BioSpace, 22 April 2026; STAT News, 24 April 2026; Petrie-Flom Centre, Harvard Law School, 18 April 2026.
Hypothesis Next explores the intersection of science, health, and the forces that shape social and financial capital. If this added value, please consider donating to the Open Medicine Foundation, the world’s leading nonprofit funding research into ME/CFS and Long Covid. A cause close to my heart. Visit OMF.ngo to donate or learn more.